Medical Disclaimer: This is educational content only, not medical advice. Consult a licensed healthcare provider for diagnosis/treatment. Information based on sources like WHO/CDC guidelines (last reviewed: 2026-02-13).
Cardiomyopathies Clinical Types Diagnosis and Management Guide
Frequently Asked Questions
What are cardiomyopathies?
Cardiomyopathies are a group of diseases of the heart muscle characterized by structural and functional abnormalities of the myocardium that are not explained by coronary artery disease, hypertension, valvular disease, or congenital heart disease.
What are the main types of cardiomyopathies?
The main types include dilated cardiomyopathy, hypertrophic cardiomyopathy, restrictive cardiomyopathy, arrhythmogenic right ventricular cardiomyopathy, stress-induced (Takotsubo) cardiomyopathy, and left ventricular non-compaction.
What is dilated cardiomyopathy?
Dilated cardiomyopathy is characterized by dilation of one or both ventricles with reduced systolic function, leading to heart failure, arrhythmias, and thromboembolic complications.
What causes dilated cardiomyopathy?
Causes include genetic mutations, viral myocarditis, alcohol abuse, chemotherapeutic drugs, peripartum state, metabolic disorders, nutritional deficiencies, and idiopathic causes.
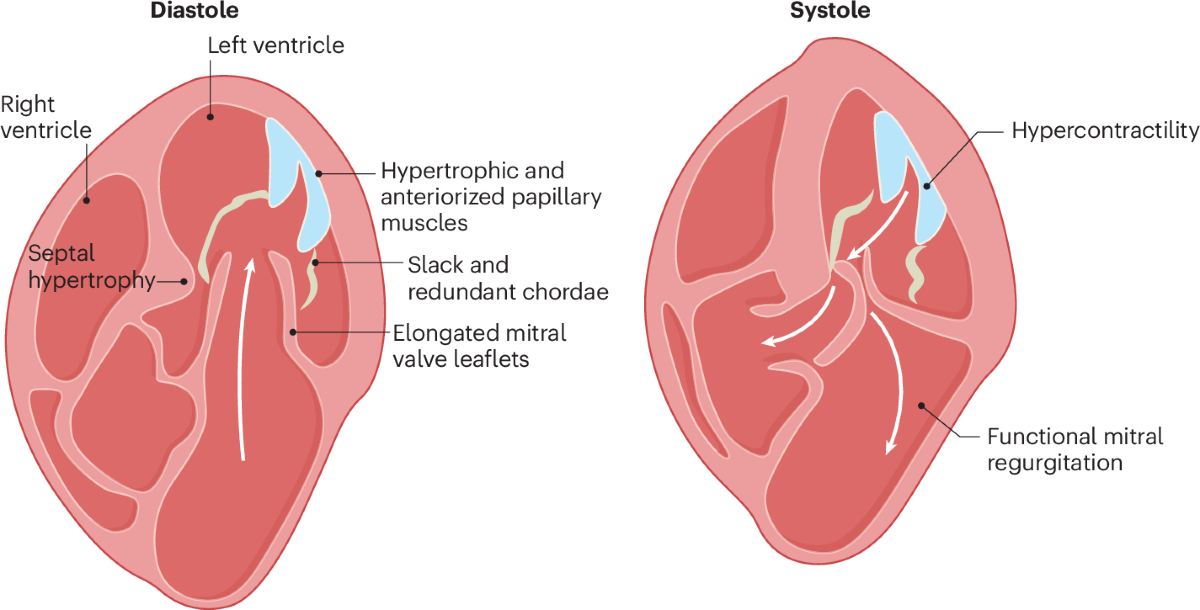
What is hypertrophic cardiomyopathy?
Hypertrophic cardiomyopathy is a genetic disorder characterized by unexplained left ventricular hypertrophy, often involving the interventricular septum, with diastolic dysfunction and possible left ventricular outflow tract obstruction.
Why is hypertrophic cardiomyopathy associated with sudden cardiac death?
Sudden cardiac death occurs due to malignant ventricular arrhythmias, myocardial disarray, ischemia, and dynamic left ventricular outflow tract obstruction, particularly during exertion.
What is restrictive cardiomyopathy?
Restrictive cardiomyopathy is characterized by impaired ventricular filling due to stiff ventricular walls with preserved or near-normal systolic function, leading to predominant diastolic heart failure.
What are common causes of restrictive cardiomyopathy?
Common causes include amyloidosis, sarcoidosis, hemochromatosis, radiation-induced fibrosis, endomyocardial fibrosis, and other infiltrative or storage disorders.
What is arrhythmogenic right ventricular cardiomyopathy?
Arrhythmogenic right ventricular cardiomyopathy is a genetic disorder involving fibrofatty replacement of right ventricular myocardium, leading to ventricular arrhythmias, syncope, and sudden cardiac death.
What are the hallmark ECG findings in ARVC?
Characteristic ECG findings include T-wave inversion in leads V1–V3, epsilon waves, ventricular arrhythmias with left bundle branch block morphology, and prolonged QRS duration in right precordial leads.
What is Takotsubo cardiomyopathy?
Takotsubo cardiomyopathy is a transient stress-induced cardiomyopathy characterized by acute reversible left ventricular systolic dysfunction, often following emotional or physical stress, with normal coronary arteries.
How is left ventricular non-compaction diagnosed?
Left ventricular non-compaction is diagnosed using echocardiography or cardiac MRI showing excessive trabeculations, deep intertrabecular recesses, and a high non-compacted to compacted myocardium ratio.
What investigations are essential in suspected cardiomyopathy?
Essential investigations include ECG, echocardiography, cardiac MRI, Holter monitoring, laboratory tests including BNP, genetic testing in selected cases, and endomyocardial biopsy when indicated.
What are the common complications of cardiomyopathies?
Common complications include heart failure, atrial and ventricular arrhythmias, thromboembolism, stroke, and sudden cardiac death.
How are cardiomyopathies generally managed?
Management includes guideline-directed medical therapy for heart failure, treatment of underlying causes, arrhythmia management, anticoagulation when indicated, device therapy such as ICD or CRT, and heart transplantation in end-stage disease.
When is an implantable cardioverter-defibrillator indicated in cardiomyopathies?
ICD is indicated in patients with reduced ejection fraction (≤35%) despite optimal therapy, survivors of cardiac arrest, sustained ventricular tachyarrhythmias, and high-risk hypertrophic or arrhythmogenic cardiomyopathy patients.
Can cardiomyopathies be reversed?
Some cardiomyopathies such as alcohol-induced, peripartum, tachycardia-induced, and stress-induced cardiomyopathies can partially or completely reverse with appropriate treatment and removal of the underlying cause.
How do cardiomyopathies differ from ischemic heart disease?
Cardiomyopathies primarily involve intrinsic myocardial disease, whereas ischemic heart disease results from coronary artery obstruction leading to myocardial ischemia or infarction.
What is the prognosis of cardiomyopathies?
Prognosis varies widely depending on the type, severity, genetic factors, response to therapy, and risk of arrhythmias, ranging from near-normal life expectancy to progressive heart failure requiring transplantation.
Is genetic screening important in cardiomyopathies?
Yes, genetic screening is important in inherited cardiomyopathies such as hypertrophic and arrhythmogenic cardiomyopathy to identify affected family members and guide surveillance and preventive strategies.
MCQ Test - Cardiomyopathies Clinical Types Diagnosis and Management Guide
Progress:
0/15
Time: 00:00
Test Results
0%
0/15
0
Correct Answers
0
Wrong Answers
00:00
Time Taken
0
Skipped