Cardiomyopathies – Comprehensive Clinical Reference
> Definition
> Cardiomyopathies are a heterogeneous group of primary myocardial disorders in which the heart muscle is structurally and functionally abnormal in the absence of coronary artery disease, hypertension, valvular disease, or congenital heart disease sufficient to explain the phenotype.
Classification (WHO / AHA / ESC – Integrated Clinical Approach)
- Dilated Cardiomyopathy (DCM)
- Hypertrophic Cardiomyopathy (HCM)
- Restrictive Cardiomyopathy (RCM)
- Arrhythmogenic Right Ventricular Cardiomyopathy (ARVC)
- Unclassified Cardiomyopathies
* Left ventricular non-compaction (LVNC)
* Takotsubo (stress-induced) cardiomyopathy
- Secondary Cardiomyopathies
* Ischemic, hypertensive, valvular, infiltrative, metabolic, toxic, inflammatory
1. Dilated Cardiomyopathy (DCM)
Definition
Progressive ventricular dilation and systolic dysfunction (LVEF <40%) involving one or both ventricles.
Etiology
- Idiopathic / genetic (TTN, LMNA mutations)
- Post-viral myocarditis
- Alcohol, cocaine, chemotherapy (doxorubicin)
- Peripartum cardiomyopathy
- Endocrine: hypothyroidism, diabetes
- Nutritional: thiamine, selenium deficiency
Pathophysiology
Myocyte injury → remodeling → ventricular dilation → reduced contractility → neurohormonal activation → heart failure.
Clinical Features
- Exertional dyspnea, orthopnea, PND
- Fatigue, pedal edema
- S3 gallop
- Functional MR/TR
- Thromboembolism, ventricular arrhythmias
Investigations
- ECG: LBBB, atrial fibrillation
- Echo: Dilated LV, ↓ EF, global hypokinesia
- Cardiac MRI: Fibrosis (LGE)
- BNP/NT-proBNP
- Endomyocardial biopsy (selected cases)
Management
A. Guideline-Directed Medical Therapy (HFrEF)
- ACEI / ARNI
- β-blocker (carvedilol, metoprolol)
- Mineralocorticoid antagonist
- SGLT2 inhibitor
- Diuretics (symptomatic relief)
B. Device Therapy
- ICD: EF ≤35% after 3 months therapy
- CRT: LBBB, QRS ≥150 ms
C. Advanced
- LVAD
- Heart transplantation
Prognosis
Variable; improved with modern therapy.
2. Hypertrophic Cardiomyopathy (HCM)
Definition
Genetic myocardial disease characterized by unexplained LV hypertrophy (≥15 mm) with or without LVOT obstruction.
Genetics
Autosomal dominant
Common mutations: β-myosin heavy chain, myosin-binding protein C
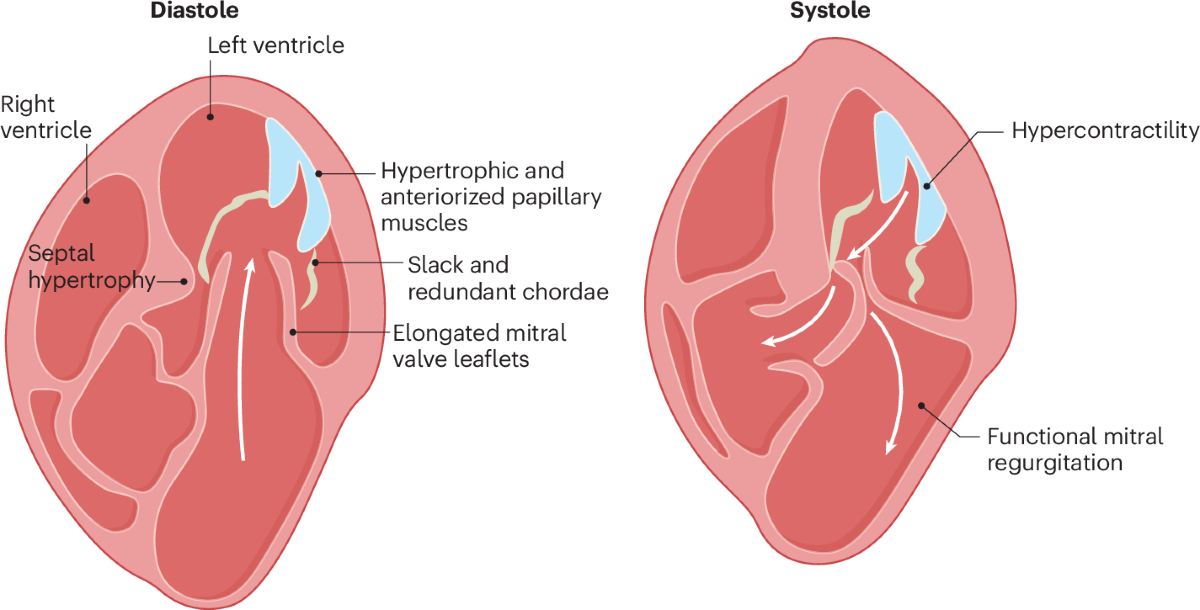
Pathophysiology
Myocyte disarray → diastolic dysfunction → dynamic LVOT obstruction → ischemia → arrhythmias.
Clinical Features
- Dyspnea, angina
- Syncope or presyncope
- Sudden cardiac death (young athletes)
- Harsh systolic murmur ↑ with Valsalva
Investigations
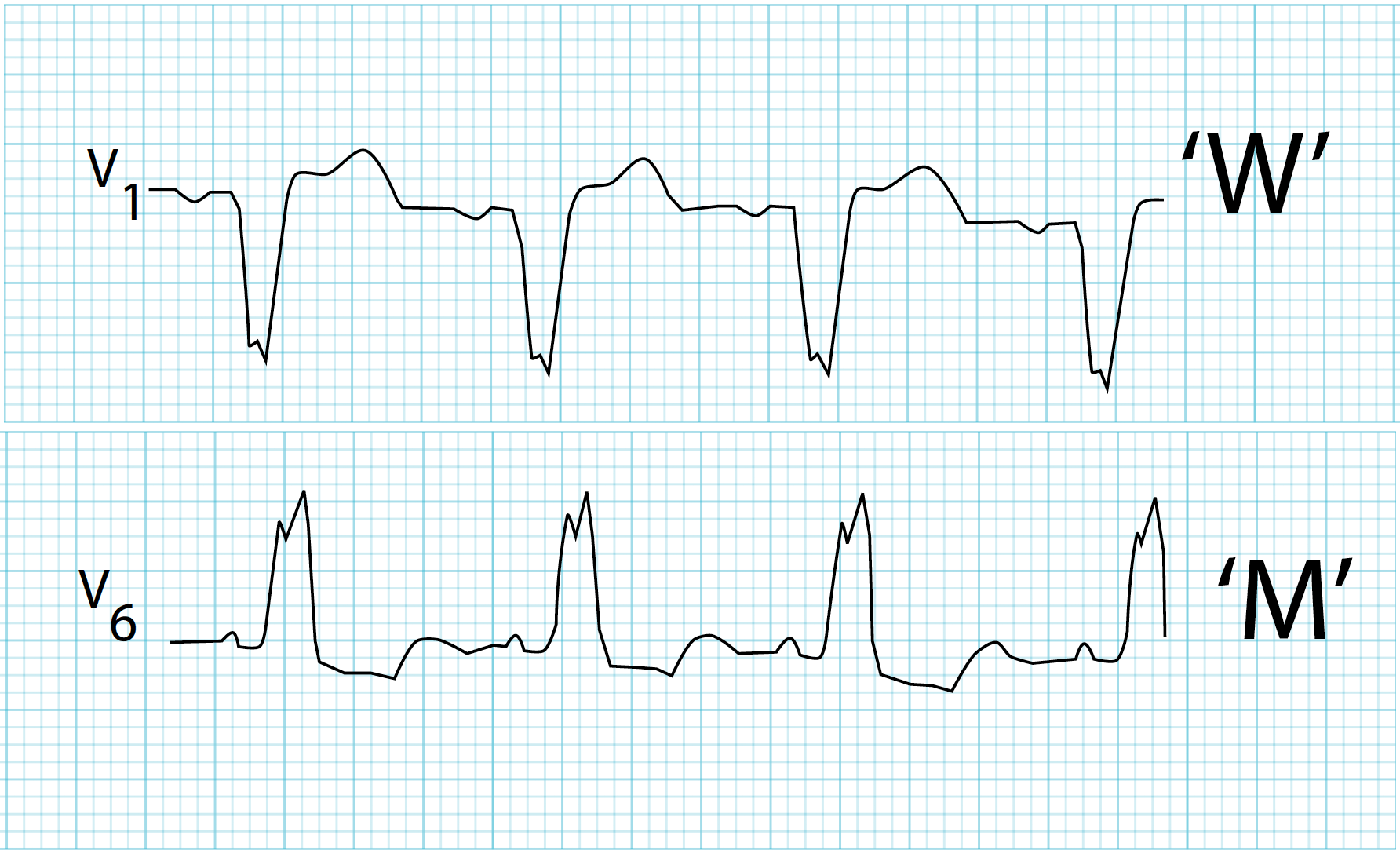
- ECG: LVH, deep Q waves
- Echo: Asymmetric septal hypertrophy, SAM of mitral valve
- MRI: Fibrosis (risk stratification)
- Genetic testing (family screening)
Management
A. Medical
- β-blockers (first line)
- Verapamil / diltiazem
- Disopyramide (LVOT obstruction)
B. Invasive
- Septal myectomy
- Alcohol septal ablation
C. Sudden Death Prevention
- ICD if:
* Family history of SCD
* Syncope
* Wall thickness ≥30 mm
* NSVT on Holter
Contraindicated
- Nitrates
- Diuretics (excess)
- Digoxin
3. Restrictive Cardiomyopathy (RCM)
Definition
Impaired ventricular filling with normal or near-normal systolic function and non-dilated ventricles.
Causes
- Amyloidosis
- Sarcoidosis
- Hemochromatosis
- Endomyocardial fibrosis
- Radiation-induced
Pathophysiology
Rigid myocardium → diastolic dysfunction → ↑ atrial pressures.
Clinical Features
- Right-sided heart failure predominance
- Ascites, hepatomegaly
- Preserved EF
- Atrial fibrillation common
Investigations
- Echo: Biatrial enlargement, restrictive filling pattern
- MRI: Infiltration
- Biopsy: Diagnostic (amyloid)
Management
- Treat underlying cause
- Diuretics cautiously
- Rate control in AF
- Transplant (selected)
4. Arrhythmogenic Right Ventricular Cardiomyopathy (ARVC)
Definition
Genetic cardiomyopathy with fibrofatty replacement of RV myocardium causing ventricular arrhythmias.
Genetics
Desmosomal protein mutations (PKP2)
Clinical Features
- Palpitations
- Syncope
- Sudden cardiac death
- VT with LBBB morphology
Investigations
- ECG: T-wave inversion V1–V3, epsilon wave
- MRI: RV dilation, fatty infiltration
- Holter: Ventricular ectopy
Management
- Avoid competitive sports
- β-blockers
- Antiarrhythmics
- ICD (high-risk)
5. Takotsubo (Stress-Induced) Cardiomyopathy
Definition
Transient LV systolic dysfunction triggered by emotional or physical stress.
Key Features
- Mimics ACS
- Normal coronaries
- Apical ballooning
- Reversible
Management
Supportive:
- β-blockers
- ACE inhibitors
- Treat triggers
6. Left Ventricular Non-Compaction (LVNC)
Definition
Failure of myocardial compaction → prominent trabeculations and deep recesses.
Complications
- Heart failure
- Thromboembolism
- Ventricular arrhythmias
Management
- HF therapy
- Anticoagulation
- ICD if indicated
Differential Diagnosis of Cardiomyopathies
- Ischemic heart disease
- Valvular heart disease
- Hypertensive heart disease
- Constrictive pericarditis
General Investigations Across Cardiomyopathies
- ECG
- Echocardiography (cornerstone)
- Cardiac MRI
- Holter monitoring
- Genetic testing
- Endomyocardial biopsy (selected)
Complications
- Heart failure
- Atrial and ventricular arrhythmias
- Sudden cardiac death
- Thromboembolism
Prognosis
Depends on:
- Type
- Genetic burden
- EF
- Arrhythmia risk
- Response to therapy
Key Takeaways
- Cardiomyopathies are myocardial diseases, not secondary to loading conditions.
- Echo and MRI are diagnostic pillars.
- Sudden death prevention is critical in HCM and ARVC.
- Early recognition improves survival and quality of life.
1. Dilated Cardiomyopathy (DCM)
Case 1
A 52-year-old man with progressive dyspnea, EF 25%, normal coronaries.
Management:
- GDMT for HFrEF: ARNI/ACEI, β-blocker, MRA, SGLT2 inhibitor
- Loop diuretics for congestion
- ICD if EF ≤35% after 3 months
Case 2
DCM patient with LBBB and QRS 160 ms, NYHA III.
Management:
- Cardiac resynchronization therapy (CRT-D)
- Continue optimal HF medications
Case 3
DCM with LV apical thrombus and ischemic stroke.
Management:
- Long-term oral anticoagulation (warfarin/DOAC if appropriate)
- Avoid antiplatelet monotherapy
Case 4
Alcoholic patient with EF 30%.
Management:
- Absolute alcohol abstinence
- GDMT for HF
- EF reassessment after 3–6 months
Case 5
End-stage DCM refractory to therapy.
Management:
- Heart transplantation
- LVAD as bridge or destination therapy
2. Hypertrophic Cardiomyopathy (HCM)
Case 6
Young patient with dyspnea, systolic murmur ↑ on Valsalva.
Management:
- β-blockers first line
- Avoid nitrates, diuretics excess, digoxin
Case 7
HCM with syncope and family history of sudden death.
Management:
- ICD implantation (primary prevention)
Case 8
Severe LVOT obstruction despite drugs.
Management:
- Septal myectomy (gold standard)
- Alcohol septal ablation if not surgical candidate
Case 9
HCM patient develops AF with rapid ventricular rate.
Management:
- Rate control (β-blocker)
- Mandatory anticoagulation regardless of CHA₂DS₂-VASc
Case 10
Asymptomatic first-degree relative of HCM patient.
Management:
- Genetic counseling and screening
- Periodic ECG + echocardiography
3. Restrictive Cardiomyopathy (RCM)
Case 11
Patient with ascites, hepatomegaly, preserved EF, biatrial enlargement.
Management:
- Treat underlying cause
- Cautious diuretics
- Rate control in AF
Case 12
Amyloidosis with low-voltage ECG and thick LV walls.
Management:
- Confirm with biopsy/MRI
- Diuretics
- Disease-specific therapy (e.g., tafamidis in ATTR)
Case 13
RCM misdiagnosed as constrictive pericarditis.
Management:
- Cardiac MRI for differentiation
- Avoid unnecessary pericardiectomy
Case 14
RCM with recurrent AF causing hypotension.
Management:
- Aggressive rate control
- Early anticoagulation
- Consider rhythm control if feasible
4. Arrhythmogenic Right Ventricular Cardiomyopathy (ARVC)
Case 15
Young athlete with syncope, VT, epsilon waves.
Management:
- ICD implantation
- β-blockers / antiarrhythmics
- Strict avoidance of competitive sports
Case 16
ARVC with frequent VT despite β-blockers.
Management:
- Add amiodarone or sotalol
- Catheter ablation for recurrent VT
5. Takotsubo (Stress-Induced) Cardiomyopathy
Case 17
Post-emotional stress, ACS-like presentation, normal coronaries.
Management:
- Supportive care
- β-blocker + ACEI
- Recovery expected in weeks
Case 18
Takotsubo with cardiogenic shock.
Management:
- Avoid inotropes if LVOT obstruction
- Mechanical support if needed
- Treat trigger
6. Left Ventricular Non-Compaction (LVNC)
Case 19
Young adult with HF and prominent trabeculations.
Management:
- HF guideline therapy
- Anticoagulation if EF low or AF
- ICD if arrhythmia risk
Case 20
LVNC with recurrent embolic events.
Management:
- Lifelong anticoagulation
- Evaluate for ICD
7. Special Cardiomyopathies
Case 21
Peripartum cardiomyopathy with EF 30%.
Management:
- Standard HF therapy (ACEI postpartum)
- Avoid future pregnancy if EF not normalized
Case 22
Chemotherapy-induced cardiomyopathy.
Management:
- Stop offending agent
- Early ACEI + β-blocker
- Serial EF monitoring
Case 23
Tachycardia-induced cardiomyopathy.
Management:
- Control heart rate or rhythm
- Often fully reversible
Case 24
Sarcoidosis with ventricular arrhythmias.
Management:
- Corticosteroids
- ICD for arrhythmia prevention
Case 25
Hemochromatosis with restrictive features.
Management:
- Phlebotomy / iron chelation
- Diuretics cautiously
- Treat systemic disease
Key Exam Pearls
- DCM → systolic failure → ICD/CRT
- HCM → diastolic dysfunction → ICD saves life
- RCM → diastolic failure → treat cause
- ARVC → avoid sports + ICD
- Takotsubo → reversible
- LVNC → embolism + arrhythmia risk