ACROMEGALY
1. Definition
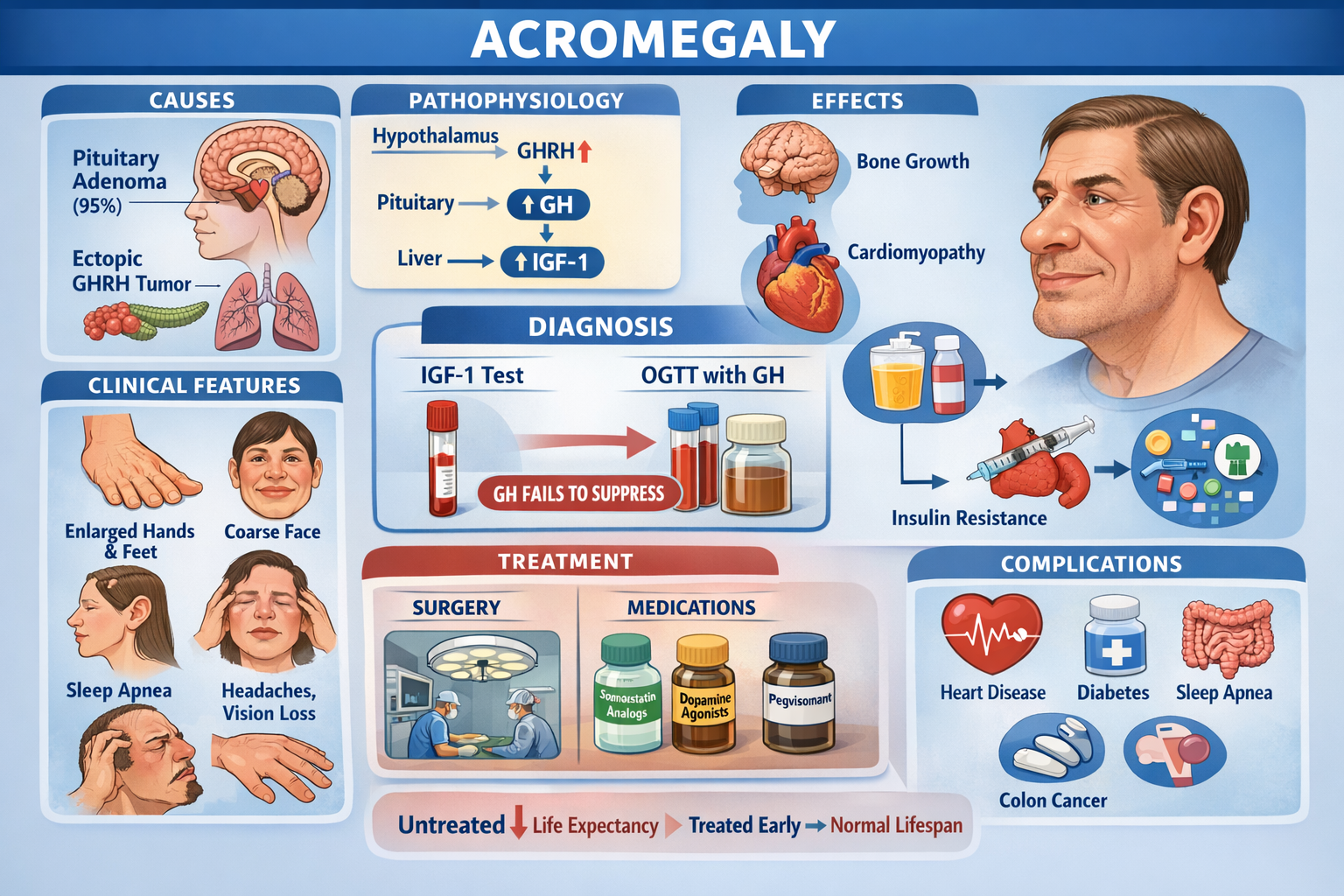
Acromegaly is a chronic endocrine disorder caused by excess growth hormone (GH) secretion after epiphyseal closure, leading to progressive enlargement of hands, feet, face, and internal organs with severe cardiometabolic and musculoskeletal complications.
(If excess GH occurs before epiphyseal closure → Gigantism)
2. Pathophysiology
Normal Physiology
Hypothalamus → GHRH ↑ → Pituitary → GH ↑ → Liver → IGF-1 ↑ → Tissue growth
Somatostatin inhibits GH.
In Acromegaly
Most commonly:
> Pituitary somatotroph adenoma → Excess GH → Excess IGF-1
IGF-1 causes:
- Bone overgrowth (mandible, hands, feet)
- Organomegaly
- Insulin resistance
- Cardiomyopathy
- Soft tissue hypertrophy
GH causes:
- Lipolysis
- Anti-insulin effect
- Sodium retention
- Increased cardiac output
3. Causes
A. Pituitary (95%)
- GH-secreting pituitary adenoma
B. Ectopic (rare)
- GHRH-secreting tumors
(Bronchial carcinoid, pancreatic NET)
4. Clinical Features
A. Skeletal
- Enlarged hands and feet (rings no longer fit)
- Prognathism (jaw protrusion)
- Frontal bossing
- Spade-like hands
- Increased shoe size
- Kyphosis
B. Soft Tissue
- Thick oily skin
- Enlarged tongue (macroglossia)
- Coarse facial features
- Deep voice
- Snoring, obstructive sleep apnea
C. Joint and Muscle
- Arthralgia
- Proximal myopathy
- Carpal tunnel syndrome
D. Cardiovascular (Major cause of death)
- Hypertension
- Cardiomegaly
- Concentric LV hypertrophy
- Diastolic dysfunction
- Heart failure
- Arrhythmias
E. Metabolic
- Diabetes mellitus
- Insulin resistance
- Hypertriglyceridemia
F. Neurological
- Headache
- Visual field defects (bitemporal hemianopia)
G. Reproductive
- Amenorrhea
- Erectile dysfunction
- Infertility
(due to ↑ prolactin or pituitary compression)
5. Investigations
A. Screening Test
Serum IGF-1 (best test)
- Always elevated
- Age-adjusted
B. Confirmatory Test
Oral Glucose Tolerance Test (OGTT)
Normal: GH suppresses to <1 ng/mL
Acromegaly: GH fails to suppress
C. Localization
- MRI pituitary with contrast
D. Complication Workup
- ECG, Echo
- Fasting glucose / HbA1c
- Lipids
- Colonoscopy (↑ colon cancer risk)
- Sleep study
6. Differential Diagnosis
- Gigantism
- Hypothyroidism (coarse face)
- Paget disease
- Pseudoacromegaly (insulin resistance syndromes)
7. Management
Goal
Normalize IGF-1 & GH, remove tumor, prevent complications
A. Surgery (First line)
Trans-sphenoidal pituitary surgery
Indication:
- Pituitary adenoma
B. Medical Therapy
Used if:
- Surgery fails
- Patient unfit
- Residual tumor
1. Somatostatin Analogs
| Drug | Octreotide, Lanreotide |
| ---- | ---------------------- |
Mechanism
Inhibit GH secretion from pituitary tumor
Dose
- Octreotide LAR 20–30 mg IM monthly
- Lanreotide 60–120 mg deep SC monthly
Adverse effects
- Gallstones
- Diarrhea
- Abdominal cramps
- Bradycardia
- Hypothyroidism
Monitoring
- IGF-1
- Gallbladder USG
- LFTs
2. Dopamine Agonists
| Drug | Cabergoline, Bromocriptine |
Mechanism
Suppress GH in some tumors (especially with ↑ prolactin)
Dose
- Cabergoline 0.5–1 mg twice weekly
Side effects
- Nausea
- Orthostatic hypotension
- Valvular heart disease (high dose)
3. GH Receptor Blocker
| Drug | Pegvisomant |
Mechanism
Blocks GH receptor → ↓ IGF-1
Dose
10–30 mg SC daily
Side effects
- LFT elevation
- Injection site reaction
C. Radiotherapy
For persistent disease after surgery + drugs
8. Complications
- Cardiomyopathy (most common cause of death)
- Hypertension
- Diabetes mellitus
- Sleep apnea
- Colon cancer
- Arthritis
- Visual loss
9. Prognosis
If untreated:
- Reduced life expectancy by 10–15 years
If treated early:
- Near-normal lifespan
10. High-Yield Exam Points
- Best screening test → IGF-1
- Best confirmatory test → OGTT with GH
- Cause → Pituitary adenoma
- Most common death → Cardiac disease
- Treatment of choice → Transsphenoidal surgery